



 You are logged in as a guest
You are logged in as a guest

 |
 |
 |
 |
The NMR spectroscopy in combination with molecular dynamics (MD) simulation is extremely powerful tool for investigation of biological molecule conformational states.
The generalized order parameter, S2, extracted from NMR and MD data, is the appropriate indicator of protein’s backbone motions in computationally feasible timescales.
Calculation of generalized order parameter S2
S2 Solver MolDynGrid Cloud Service (https://www.ncbi.nlm.nih.gov/pubmed/15915725) 
A peptide N-H bond motions is described from the NMR relaxation data using the correlation motion function (CMF) which is expressed for each residue as:
Cτ = <μtμτ+t>t
where μ(t) is the unit vector along the N-H bond direction at the time t. When the overall protein motion is not taken into account (i.e. when the coordinate system is associated with protein) the function is called as internal correlation motion function (internal CMF). To process the internal CMF with respect to time can be described either by the simple model–free approach:
Ct=S2+1-S2exp-tτ
оr by the extended model–free approach:
Ct=S2+1-Sf2exp-tτf+Sf2-S2exp-tτs
On basis of the GROMACS template a program for correlation motion function calculation was written. As model system we chose molecular dynamics of EMAP II during 100 ns that was previously computed. Using the program g_rotacf from Gromacs we computed the time-dependent internal CMF for all residues of the EMAP II. Then we have calculated S2 with the simple model–free approach proposed by Dubyna V.M. and used S2 already calculated from our NMR experimental data present work, different methods were performed for N-H bond S2 order parameter calculation from MD simulation data of EMAPII and compared with results of NMR experiments
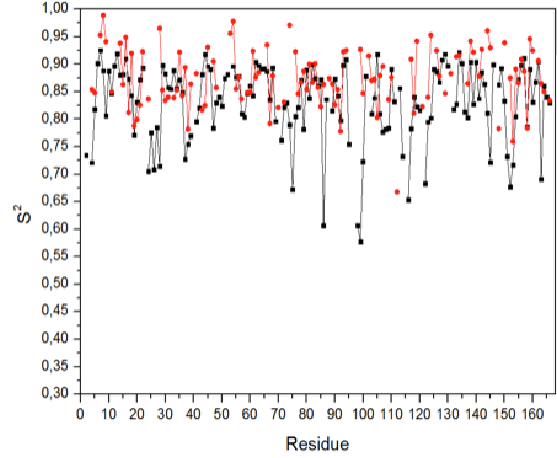
The program s2solver based on this algorithm is made and adjustable parameter influence on the calculation results was studied. The calculated order parameter values are in satisfactory agreement with those obtained by other authors using NMR.
If you use s2solver, please cite:
Kovalskyy D. B., Ivanova O. S., Dubyna V. M., Kanibolotsky D. S., Kornelyuk A. I. Generalized ordering parameter S2 as measure of the conformational flexibility of the proteins: comparison of algorithms of S2 calculation from molecular dynamics simulation data, Ukr. Biokhim. Zhurn.–2004.–V. 76, N 2.–P. 128-132.
Dubyna V.M., Kovalskyy D.B., Ivanova O.S. and Kornelyuk A.I. The improvement of the algorithm for order parameter calculation (S2) from molecular dynamics simulation using the correlation motion function, Biophysical Chemistry , Vol.123, N1, p. 25-28, 2006.